Evolution and phenotypic impact of de novo genes
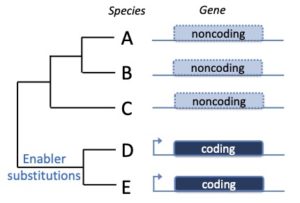
New protein-coding genes can evolve from scratch from noncoding DNA sequences. These so-called de novo genes (DNGs) have been found in a number of species and are implicated in the emergence of novel phenotypes, for instance resistance to low water temperature in artic and antartic fish. Some strategies used to identify DNGs have proven somewhat inaccurate (Casola 2018). We are developing new tools to find DNGs and to assess their phenotypic impact both computationally and experimentally.
Gene evolution in conifers

Comparative analyses of conifer genomes have shown that a large fraction of genes in these species have no detectable homologs in angiosperms or other eukaryotes. Our group is developing computational tools to determine the possible role of gene duplication and other processes in the emergence of lineage-specific genes in conifers. We have recently determined that, contraty to the nucleotide substitution rate, conifers experience a comparable frequency of gene duplication and loss compared to flowering plants (Casola and Koralewski 2018). In collaboration with other forest genomic groups across the country, we have deciphered the evolutionary dynamics of gene families in other conifer genomes (Neale et al. 2017).
Signatures of convergent evolution in chloroplast genes
The C4 photosynthetic pathway has evolved more than 60 times in flowering plants. Several key photosynthesis-related enzymes, including RuBisCO, the primary protein fixing atmospheric CO2 in plants, show signatures of molecular convergence in the form of recurrent identical amino acid replacements between C4 lineages. However, it is unclear how many proteins might share convergent amino acid replacements as a result of the transition to C4 photosynthesis. In a recent study, we tested if proteins encoded by chloroplast genes showed evidence of convergent replacements in C4 groups compared to sister C3 lineages in the PACMAD clade of grasses. Our results show that C4 lineages share significantly more convergent replacements among chloroplast proteins than their sister C3 taxa. Our findings may help developing more resilient crop varieties that integrates components of the C4 pathway.
Bark beetle genomics and convergence of the wood-boring habit
 Bark beetles are common forest pests responsible for the annual loss of millions of conifers and other trees in the United States. Dendroctonus frontalis, also known as the Southern pine beetle (SPB), is the most damaging forest pest in the southeastern United States, having caused losses estimated at more than $1.5 billion. SPB outbreaks have primarily affected loblolly pine, which forms 80% of the planted forestland and over one-half of the standing volume in the southern U.S. SPB is increasingly threatening pine forests across Central and North America and has recently colonized previously unaffected areas north of its historical range due to climate change (i.e. higher average winter temperatures). To better understand the biology and evolution of this pest, we are sequencing and analyzing the genome and transcriptome of the SPB in collaboration with Dr. Heath Blackmon, Department of Biology @TAMU and Prof. Lynne Rieske-Kinney at the University of Kentucky.
Bark beetles are common forest pests responsible for the annual loss of millions of conifers and other trees in the United States. Dendroctonus frontalis, also known as the Southern pine beetle (SPB), is the most damaging forest pest in the southeastern United States, having caused losses estimated at more than $1.5 billion. SPB outbreaks have primarily affected loblolly pine, which forms 80% of the planted forestland and over one-half of the standing volume in the southern U.S. SPB is increasingly threatening pine forests across Central and North America and has recently colonized previously unaffected areas north of its historical range due to climate change (i.e. higher average winter temperatures). To better understand the biology and evolution of this pest, we are sequencing and analyzing the genome and transcriptome of the SPB in collaboration with Dr. Heath Blackmon, Department of Biology @TAMU and Prof. Lynne Rieske-Kinney at the University of Kentucky.
Additionally, we are using comparative genomic approaches to identify gene families involved in the independent establishment of the wood-boring habit across multiple lineages of Coleoptera.
Genetic basis of drought tolerance in loblolly pine

Other topics of interest include genome size evolution, gene duplication via retroposition and horizontal transfer of transposable elements.